问题分诊室
案例分享 | 大牛综述T2T时代的基因组组装(下)
发布时间: 2024-06-03
近期,生物信息领域大神、美国丹娜-法伯癌症研究所李恒老师在Nature Reviews Genetics上发表了关于T2T基因组的综述文章《Genome assembly in the telomere-to-telomere era》,该文回顾目前在大型真核生物基因组高质量组装方面的实践,最终指向端粒到端粒(T2T)的组装。文章从6个方面对基因组T2T组装进行了总结,介绍常见T2T组装的数据类型,剖析最新的组装算法,解释评估组装的方法,讨论组装T2T基因组的挑战,为组装T2T基因组提供了自己的见解和指导。上篇文章“案例分享 | 大牛综述T2T时代的基因组组装(上)”已经在前3个方面进行了解读,本文将对最后3个方面进行解读。
核心组装算法
reads纠错
虽然PacBio HiFi和ONT双链reads很准确,但它们并非没有错误。残存的错误与基因变异混杂在一起,可能会妨碍同源单倍型或重复拷贝的分离,从而导致组装结果支离破碎。所有支持T2T的组装软件都会尝试纠正reads上的测序错误,HiCanu、verkko和hifiasm会将所有reads相互比对。对于每个reads,如果该碱基在比对到同一位置的其他重叠reads中很少出现,它们就会对该碱基进行校正。LJA在不进行错误校正的情况下构建初始装装图,将每个原始reads与组装图比对,并将高k-mer覆盖率的组装图路径作为校正后的序列。HiCanu、Verkko和LJA压缩reads中的同源多聚物,并在同源多聚物压缩空间中校正reads,而hifiasm则在原始碱基上校正,这些组装软件可以纠正大部分错误。
很有可能在很大程度上,不同组装软件对相同数据的结果差异既取决于组装算法的差异,也取决于纠错的差异,但这很难确定是哪种原因,因为组装和纠错通常是结合在一起的。如果方法开发者能将纠错步骤与组装步骤分开,将会有所帮助。
基于重叠图的组装
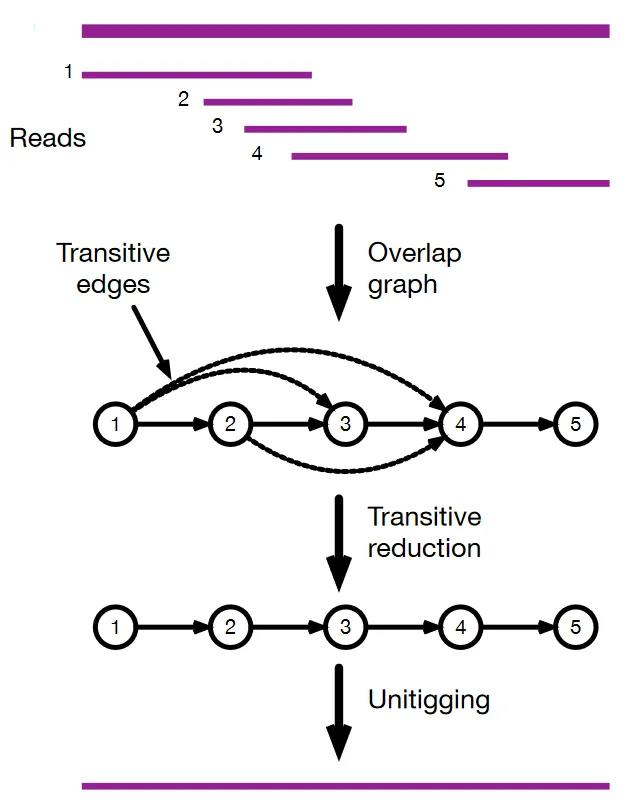
在重叠图中,每个顶点都是一个read。如果read A的后缀能与read B的前缀比对上,就会在read A和read B之间添加一条有向边(图4)。
图4 基于重叠图的组装
实际的重叠图并非如此清晰,需要进一步处理。特别是当重复长度大于重叠长度时,不同的重复拷贝之间会出现重叠(图5)。就人基因组而言,如果将重叠长度控制在几kb以下,那么约6kb LINE1重复序列之间就会有很多重叠。尽管如此,鉴于reads比LINE1长,预计唯一区域的重叠比重复重叠长。如果一条read有两个重叠(图5中read2和read3、4),较短的重叠更有可能是由重复引起的,可能会被剪切掉。同时,未修正的测序错误可能会导致额外的“尖端”(如图5中的read3)或“气泡”,这些也会在图形清理过程中去除。
图5 重复序列对组装的影响
大多数基于重叠的组装软件都遵循图4、5中的流程,HiCanu和hifiasm是针对HiFi reads优化的基于重叠的组装软件,它们与其他组装软件的不同之处在于只允许完全重叠。这一看似微小的差别正是它们区分重复拷贝(图6A)和单倍型分相(图6B)能力的主要来源。通过这种方式,它们比其他组装软件拥有更连续、更精确的结果。
图6 HiCanu和hifiasm对串联重复和二倍体样品的组装
基于de Bruijn图的组装
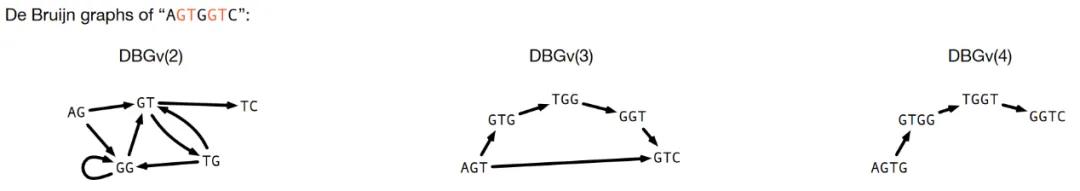
构建de Bruijn图(DBG)有两种方法:以节点为中心或以边缘为中心(图7)。在以节点为中心的图中,即DBGv(k),每个顶点都是reads中的一个k-mer,如果两个k-mer之间有k-1个碱基重叠,就会有一个边。在以边为中心的图DBGe(k)中,每条边都是reads中的一个k-mer,每个顶点都是一个 (k-1)-mer。从数学上讲,DBGe(k+1)是DBGv(k)的子图,而DBGv(k)是DBGe(k)的线图。在文献中,这两种定义都很常见,由于其与重叠图的关联性,在本综述中采用以节点为中心的观点。以节点为中心的DBG是一种重叠图,由位于顶点的k-mer和对应于k-1 bp重叠的边组成。它没有包含reads或传递边,因此比较简单。用于重叠图的策略通常也适用于DBG。
图7 以节点为中心的de Bruijn图
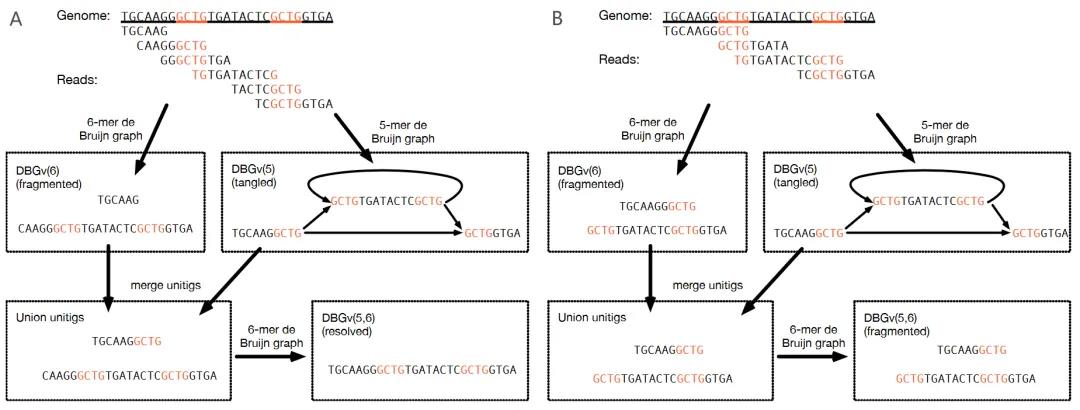
基本DBG会丢弃长度超过k-mer大小的信息,这就降低了分相和重复序列解析的能力。选择大的k来保留长距离信息可能很诱人,但使用大的k会增加在低覆盖率区域出现contig断点的几率(图8A),没有一个适用于所有情况的最佳k值。多重DBG为k-mer选择的难题提供了一个很好的解决方案。从概念上讲,多重DBG可以看作是以不同k-mer大小构建的多个DBG的合并(图8B)。它在重复区域自适应地选择大的k,在低覆盖区域选择小的k。然而,使用多重DBG并不能解决DBG中的所有歧义(图8B)。实际的组装软件会启发式地在不同的子图中使用不同的k-mer大小集。他们检索子图中使用的reads,如果新的子图更简单且保持连续,则用较长的k-mer构建的新子图替换该子图。
图8 多重DBG组装
基于minimizer的稀疏化,是现代组装软件采用的另一种技术。只在内存中保留minimizer或closed syncmers(所有k-mer的一个小子集),而不是存储reads中的每个k-mer。这一策略大大减少了内存,加快了构建速度。一种相关但不同的构造称为minimizer-space DBG(mDBG),它使用k个连续的最小化子作为 “k-mers ”来构造DBG。MetaMDBG实现了多重mDBG,这与图8A中多重DBG的概念定义非常接近。
虽然大多数短reads组装都使用DBG方法,但没有组装软件使用DBG来组装错误率大于5%的长reads,因为保留长距离信息所需的长k-mers大多是错误的。不过,有了准确的长reads,就可以纠正大部分测序错误,并使用长度超过10kb的k-mers,DBG再次成为可行的选择。Verkko和LJA是基于DBG的组装软件,其装配质量与HiCanu和 hifiasm大致相当。
整合多种数据类型
对于二倍体样本来说,只有当一个read同时包含两个位点时,才能将两个杂合位点相加。人类和许多其他物种的二倍体基因组包含许多比HiFi reads更长的区域,而且不包含任何杂合位点。我们无法仅用HiFi reads对人类基因组进行分期。同时,在过去数千年中发生的片段复制很可能在数十kb的范围内保持相同,而HiFi读数也无法解决这一问题。我们需要额外的数据类型来获得染色体长相位,并对最近的重复进行组装。
最强大的辅助数据类型是ONT超长reads,原因很简单,因为它们的读数长度很长。Verkko将超长reads与初始组装图对齐,并识别通过该图的路径。然后,它使用相同的读线程算法简化初始图,构建多个DBG。Hifiasm则使用与minigraph相似的算法进行超长reads到图的比对。它将超长reads编码为单元格序列,并在单元格空间应用基于重叠的组装算法。虽然这两种组装软件使用的算法不同,但它们都能在整合超长reads后生成节点更少、拓扑结构更线性的简化组装图。对于人类数据,新图将解决大部分复杂区域,并包含平均长度超过10Mb的分相区域。
三联体数据是通过对样本的双亲测序获得的,是全基因组分相最可靠的数据类型。有了三联体数据,就能识别只出现在其中一个亲本中的k-mer,并用这些k-mer标记组装图中线性片段的亲本来源。例如,可以将父本线性片段和未标记线性片段连接起来,从而得到长父本等位基因。verkko和hifiasm都可以使用这类信息。然而,在实践中,由于人类伦理方面的考虑,或者由于野生捕获动物的亲本无法获得,亲本数据可能难以获得。原则上,其他近亲的数据也能提供类似的信息,但这类信息很少用于组装新物种。
当三联体数据不可用时,Hi-C对于染色体尺度的分相至关重要(图3d)。Hifiasm和GFAse改写了基于参考基因组的Hi-C分相算法用于全新组装。它们检查Hi-C比对到contig的情况,如果两个contig共享匹配到相同的Hi-C片段,则在它们之间施加吸引力;如果它们的序列相似性很高,则施加排斥力。吸引力倾向于将具有相同相位的片段组合在一起,而排斥力则将同源片段推向相反的相位。Hifiasm和GFAse试图在这两种力之间找到平衡点,从而对contig进行分相。Verkko可以选择使用GFAse phasing对整个染色体进行分相。此外,如果已知并足够频繁地标记每一对同源contig,还可以使用印记甲基化标记来确定这些染色体的亲本来源。
除了分相之外,如果组装图中仍有空白,Hi-C还能在构建scaffold方面发挥关键作用(图3e)。有几种针对高质量 HiFi组装进行优化了的搭建scaffold软件,VGP和DToL都使用YaHS搭建scaffold。
抛光是指为提高contig序列的碱基准确性而实施的过程。对于纯合基因组,错误的contig碱基不会得到read比对或read k-mers的支持,因此可以被识别出来。对于二倍体的单倍型分辨率的组装,可以通过单倍型基因组之间比对或reads与基因组比对得到的变异识别出错误的contig。重要的是,为未分相组装开发的抛光工具通常不用于单倍型解析装配,因为它们会混合来自不同单倍型和重复拷贝的reads,从而降低contig碱基的准确性。
评估序列组装
要想真正实现T2T的组装,就必须既能覆盖每条染色体的全部,不留缝隙,又不包含大规模的组装错误。在得出T2T的结论之前,必须严格评估组装的质量。
基本指标
为了获得对组装的第一印象,会计算组装大小(所有contig长度的总和)以及N50(N50的定义是contig长度不短于长度覆盖组装的一半长度的数字)。对于二倍体的常染色体,我们希望一对组装(图3c)或染色体分相组装(图3e)中的两个组装序列具有相似的大小。一对不平衡的常染色体组装可能表明不完全分相,可能需要手动调整参数或进行整理。当然,在异形配子中,性染色体很可能具有不同的大小。物种内部也会出现其它倍性差异,例如体细胞染色体缺失或减少。
评估基因的完整性
BUSCO仍是评估组装完整性的黄金标准。一个不太完整的组装会有更多的蛋白质未比对,需要注意的是,BUSCO可能会低估大型基因组的完整性,这是因为它在将蛋白质序列与基因组准确比对方面存在局限性。例如,在有注释的人类基因上运行 BUSCO会得出99.2%的完整性,但在GRCh38基因组BUSCO的完整性仅为95.7%。
minimap2软件包中的 “asmgene”工具是BUSCO的替代工具,在有高质量参考基因组的情况下也能解决完整性低的问题。该工具基于cDNA与参考基因组的比对来识别单拷贝基因,并额外评估参考基因组中的多拷贝基因是否与目标基因组中的多拷贝基因组装在一起。噪声read组装软件可能会组装出单拷贝基因,但往往会漏掉多拷贝基因。
虽然基因具有重要的生物学意义,但它们只占基因组的一小部分。基于基因的评估,尤其是基于单拷贝基因的评估,会遗漏复杂的基因组区域,因此应辅以其他评估方法。
基于K-mer的评估
如果read覆盖率均匀且reads组装完美,预计组装中k-mer的数量与其在reads中的数量成正比。一个k-mer在组装中出现的频率高于在reads中出现的频率,表明组装中存在错误的重复,而一个k-mer在reads中出现的频率高而在组装中出现的频率低,则表明序列缺失。KAT是一款功能强大的工具,可利用这些简单的观察结果来评估组装。
现在通常的做法是使用k-mers来估计contig序列的碱基准确性,通常用Phred标度作为质量值来衡量,该方法的工作原理是计算reads中不存在的contig k-mers的比例,分数越高表明QV越低。目前,Merqury和yak可以实现该分析。值得注意的是,由于质量值的估算取决于输入reads的质量和深度,因此基于不同输入reads的质量值估算并不具有严格的可比性,要得出一个物种的质量值高于另一个物种的质量值的结论并不容易。此外,使用同一样本中不同技术产生的reads来衡量质量值也很常见。虽然这种方法有助于减少系统性测序误差的影响,但如果有无reads覆盖区域,则可能会低估质量值。当有三联体数据时,Merqury和yak也可以使用父本特异性k-mers来评估组装的分相准确性。
基于比对的评估
理想情况下,当我们将reads与它们组装的基因组对齐时,我们希望每个contig有着均匀覆盖率,在较长区域内过低或过高的覆盖率将表明潜在的组装错误。我们还希望contig能够得到reads碱基的良好支持。当用reads与组装基因组比对得到变异时,获得的小变异暗示contig一致性错误,而聚类的杂合变异可能是由折叠的片段重复引起的。这些信号在评估纯合CHM13基因组中起着至关重要的作用。对于二倍体基因组,我们可以合并两个单倍型组装,并将reads比对到合并体中,应该观察到类似的信号。Flagger和Inspector是面向用户的评估工具,基于reads比对组装基因组的方法。
对于像CHM13这样近乎完美的样本,可以将现有的组装作为基本事实来评估自动组装。QUAST是用于此目的的流行工具,这种基于组装与组装比对的方法对于组装软件开发人员调整组装算法非常有用,但不适用于新物种,也不适用于 "真实 "组装和评估组装来自不同菌株或样本的情况。如果真实的组装结果和评估的组装结果来自不同的菌株或样本,QUAST就不能很好地将两个样本之间的真正差异与组装错误区分开来。在复杂区域中更完整的装配会包含更多的结构变异,错误率似乎也会更高。例如,如果将人类参考基因组GRCh38作为基本真相来评估T2T-CHM13组装,QUAST将报告超过20,000个错误组装。
从头序列组装的挑战
尽管取得了上述进展,但从头序列组装并不是一个已解决的问题。虽然所有主流组装软件都建立在1995年建立的基本组装算法之上,但它们严重依赖没有坚实理论基础的手动调整启发式算法。受限于实际数据的特征,它们无法解析基因组中最复杂的区域。它们在多倍体基因组或更复杂的病例(例如具有异质性大规模结构变异的癌症基因组)中也表现不佳。
理论挑战
在重叠图和de Bruijn图这两种组装模型中,每种模型都有自己的注意事项。在构建重叠图时,如果一条read包含在其他更长的reads中,它将会被丢弃。当reads长度可变时,这一看似简单的步骤可能会导致组装间隙。这种组装间隙并不常见,但由于现代组装是高度连续的,因此由被包含reads引起的额外组装间隙是显而易见的。消除被包含reads是基于重叠的组装算法的致命弱点,仍然是一个有待解决的关键问题。
领先的DBG组装软件,包括用于短读数的SPAdes和用于长读数的Verkko和LJA,都使用多重DBG,这与教科书中描述的基本DBG不同。虽然理论上已经研究了从所有输入reads的固定k-mer集构建多重DBG,但实际的DBG组装软件并不使用这些算法,因为一个k-mer集可能无法在所有子图中达到最佳效果。实际的DBG组装软件采用启发式方法,在图的连续性和复杂性之间徘徊。我们将拭目以待,看能否开发出一种新的组装模型,将不同的长度尺度更顺畅地结合在一起。
实际挑战
由于Hi-C只提供contig之间的相对相位信息,因此使用Hi-C作为长程数据进行组装比使用三联体数据进行组装更加困难。如果没有三联体数据,目前的组装软件可能难以解决复杂的情况。此外,它们可能无法将异源染色体样本中的性染色体清晰地分离出来。不过,通过检查Hi-C比对,VGP和DToL往往可以发现这些问题,并手动解决它们。这表明,Hi-C数据的使用还有进一步改进的空间,或许可以使用机器学习方法。
目前的T2T策略强调使用准确率远高于99%的长reads。虽然Canu、Flye和Shasta可以用错误率大于5%的ONT reads,组装出拥有数十Mb contig的CHM13基因组,但它们产生了更多的结构性错误:在完成的T2T-CHM13基因组中,24-75%的多拷贝基因被这些组装软件错误组装,而HiCanu或hifiasm的错误率仅为1%,它们还正确重建了更多的细菌人工染色体序列。一般来说,噪声长reads的组装并不与准确长reads的组装竞争,不过,ONT正在迅速提高单条reads的准确性,我们认为有可能为这些新的改进型reads设计纠错策略,从而允许使用当前的或经过调整的需要精确匹配的长reads组装算法。如果这种方法行之有效,那么就有望通过单一数据类型实现准确的T2T组装,从而大大简化高质量基因组组装工作。
在实践中,实验挑战往往比计算挑战对组装质量的影响更大。标准的长片段生成方案需要大量的DNA(>1μg),而从小型生物体或临床样本中很难或根本不可能获得这些 DNA。PacBio有低至0.1μg的无扩增低输入文库方案,但低于此阈值时,就必须进行全基因组扩增,这会带来覆盖偏差和丢失。长reads也存在覆盖偏差。例如,PacBio HiFi reads很难对富含GA的长重复序列进行测序,最近的一项研究表明,这导致了超过25%的组装缺口,而据报道,ONT reads在端粒序列方面也有问题。
超越二倍体样品
虽然最近在二倍体基因组的T2T组装方面取得了很好的进展,但对多倍体基因组还没有令人满意的解决方案。最近有两种四倍体马铃薯的单倍型分相组装,使用单细胞测序数据或遗传图谱进行分相,但这些方法不能大规模部署。我们更愿意使用通用数据类型推导多倍体组装:HiFi长reads、超长reads和Hi-C数据。
癌症基因组在某种程度上是多倍体,染色体之间或染色体内部的倍性各不相同,因此更难组装。除了癌症之外,能够组装宏基因组样本也是有益的,因为宏基因组包含大量物种,通常是微生物,而且相对丰度非常不同。宏基因组也可视为多倍体基因组,其倍性变异更大。不过,在组装宏基因组样本时,与多倍体基因组组装相比,门槛较低:例如,通常不会出现高度相似的基因组。有一些专门的宏基因组组装软件,如MetaFlye、hifiasm-meta和metaMDBG,可以从深度测序的宏基因组样本中重建多达几百个封闭细菌基因组,尽管16S或k-mer图谱显示存在缺失物种。要实现完整的宏基因组组装,还有很长的路要走。
参考文献
Li, H., Durbin, R. Genome assembly in the telomere-to-telomere era. Nat Rev Genet (2024). #
康普森农业长期致力于动植物基因组、泛基因组和T2T基因组组装研究,拥有丰富的项目方案设计及分析经验,涵盖家禽、家畜、粮食作物、园艺作物、花卉林木、水产等多样性物种,将为科研工作者提供全面的三代测序、基因组组装和群体重测序等方面的专业技术服务。
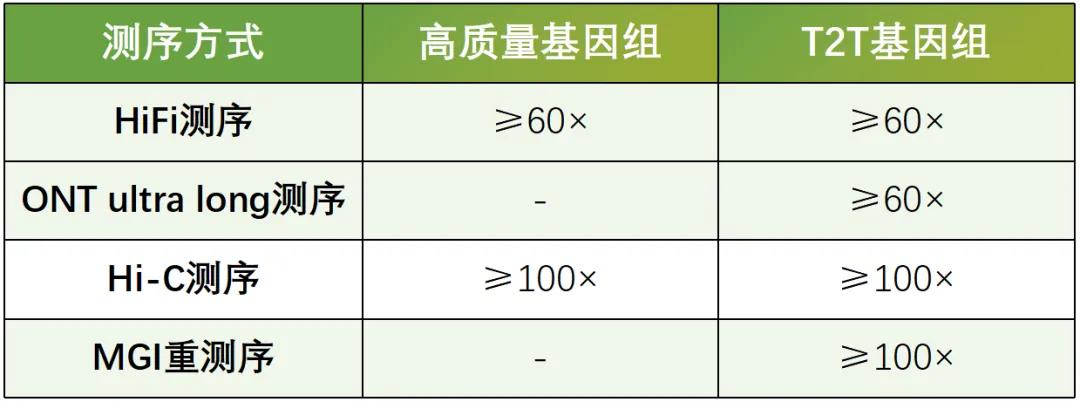
康普森基因组组装方案
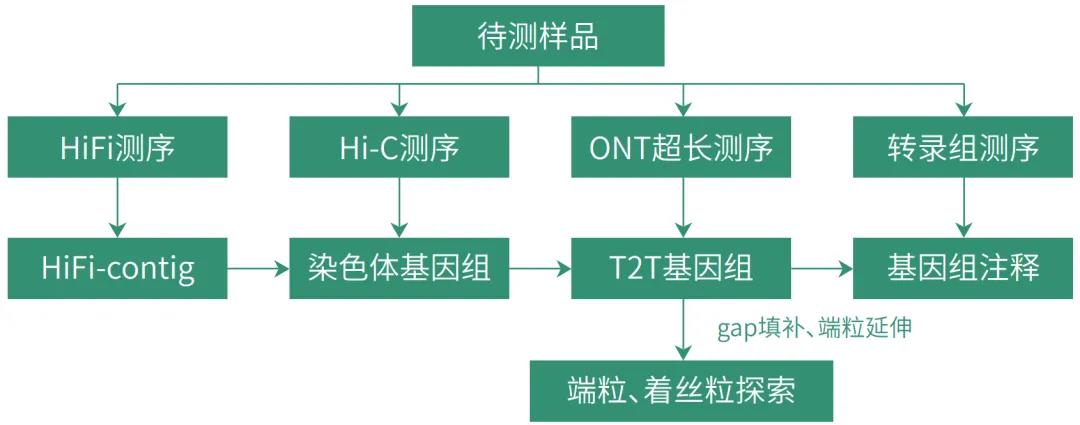
针对T2T基因组组装,康普森推荐使用高准确性的HiFi reads和高连续性的ONT超长reads和Hi-C reads共同分析。
康普森采用多种策略进行T2T组装,首选使用HiFi reads搭建基因组框架,然后使用Hi-C数据将初组装序列挂载到染色体水平,而后再利用ONT超长reads对染色体中gap进行填补,同时对端粒序列进行延伸组装,最终获得T2T基因组。
天津:18710280840/022-24986099
北京:400 1869 509
邮箱:marketing@kangpusen.com
地址:北京市昌平区中关村生命科学园生命园路4号院4号楼7层
图文来源:北京康普森农业科技有限公司









 下载app
下载app