问题分诊室
技术分享 | 使用BLAST评估二代测序数据是否污染
发布时间: 2024-05-20
由于NGS测序时混库的原因,我们得到的数据有被其他物种污染的可能,所以当我们拿到测序数据时,如何确定我们的数据是否被污染呢?
下面我们通过BLAST软件的比对结果,来检查测序数据fastq 是否被污染,以及污染来源、所占比例。
BLAST (Basic Local Alignment Search Tool) 是我们常用的短序列比对工具,直接输入fasta格式的序列文件就可进行比对。
## 下载Blastwget -cftp://#/blast/executables/blast+/LATEST/ncbi-blast-2.15.0+-x64-linux.tar.gz## 解压tar -xvzf ncbi-blast-2.15.0+-x64-linux.tar.gz
BLAST序列比对的工具共有5种,大家可以根据自己序列比对的类型进行选择。
· blastn:将核苷酸序列比对至核苷酸数据库。
· blastp:将氨基酸序列比对至氨基酸数据库。
· blastx:将核苷酸序列比对至氨基酸数据库。
· tblastn:将氨基酸序列比对至核苷酸数据库。比对时,将输入的氨基酸序列与数据库中核苷酸序列翻译后的氨基酸序列逐一比对。
· tblastx:将核苷酸序列比对至核苷酸数据库。与blastn的区别是比对时,输入的核苷酸序列与数据库中的核苷酸序列都先翻译为氨基酸序列,而后再进行逐一比对。
本次分析使用的blastn软件。
本次示例数据来源于2个水稻样本的fq文件。
nt/nr fasta下载与建库
从NCBI(ftp:#)下载nt、nr fasta文件。在进行序列比对前,我们首先需要构建数据库。
wget ftp://#/blast/db/FASTA/nt.gzwget ftp://#/blast/db/FASTA/nr.gztar -xzvf nr.gztar -xzvf nt.gzmkdir nr_dbmkdir nt_db## 构建数据库makeblastdb -in nr -dbtype prot -title nr -parse_seqids -out ./nr_db/nr -logfile make_nr.logmakeblastdb -in nt -dbtype nucl -title nt -parse_seqids -out ./nt_db/nt -logfile make_nt.log
-in:构建数据库所用的序列文件。
-dbtype:数据库类型。构建的数据库是核苷酸数据库时,dbtype设置为nucl,数据库是氨基酸数据库时,dbtype设置为prot。
-out:数据库名称。
或者直接使用NCBI处理好的db:
wget -c ftp://#/blast/db/nt*wget -c ftp://#/blast/db/nr*
NCBI Taxonomy 数据库下载
wget -c https://#/pub/taxonomy/accession2taxid/nucl_gb.accession2taxid.gzwget -c https://#/pub/taxonomy/taxdump.tar.gz
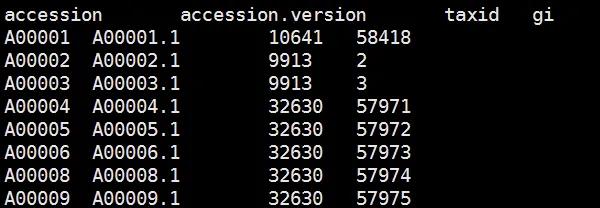
nucl_gb.accession2taxid 数据库格式如下:
第一列Accession :序列标识码
第一列Accession.version : 带版本号的序列标识码
第三列:序列的taxid 号,即物种分类号。如 Homo sapiens 的是9606.
第四列:序列的gi号
这个文件比较大,我们后边只用三四列信息,所以可以把这个库处理一下,只留下第3,4列。
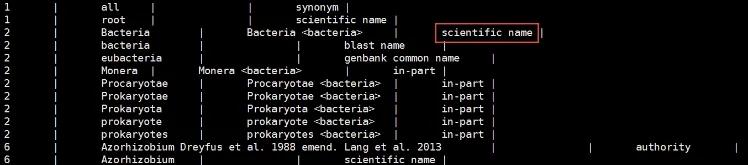
cut -f 3,4 nucl_gb.accession2taxid > cutted_nucl_gb.accession2taxidtaxdump.tar.gz 解压后会有7个库,我们用 names.dmp ,其中包含物种的taxid号与物种学名。对应的格式如下:
第1列为物种的taxid号。
第2列为物种名称。
我们后面会选择scientific name 对应的物种学名。
fa文件提取
从样品fastq文件中随机抽取部分序列,并将fastq转换为fasta格式。
序列比对
转化之后的fasta文件与NT数据库进行比对。命令如下:
blastn -num_threads 32 -max_hsps 1 -max_target_seqs 1 -evalue 1e-05 -db nt -outfmt "7 qseqid sseqid evalue pident ppos length mismatch gapopen qstart qend sstart send bitscore staxid sscinames stitle" -query *.fa -out *.outblastn结果详解
query_id refer_id identity alignment_length mismatches gap_openingsq.start q.end s.start s.end e-value bit_scoreBA000030.4 CP023202.1 78.25 1209 226 26 1581317004 7017078 7018266 0 741BA000030.4 CP016438.1 75.15 4363 967 102 2609730398 6410499 6414805 0 1943BA000030.4 CP023692.1 71.89 3163 704 141 2609829169 4386693 4389761 0 745BA000030.4 CP033073.1 76.29 3108 642 77 2610529169 2959523 2956468 0 1568BA000030.4 CP060828.1 74.8 2936 661 63 2615729058 5940668 5937778 0 1249BA000030.4 CP016279.1 76.88 3032 631 63 2616829170 9144413 9147403 0 1652BA000030.4 CP034539.1 73.95 2349 521 77 2624128542 2852251 2849947 0 863BA000030.4 CP033073.1 84.84 2164 307 16 2927031427 3675813 3677961 0 2159
总共对应12列结果,每一列的含义如下:
query id:查询序列ID标识;
refer id:参考序列ID标识;
identity (%):序列比对的一致性百分比;
alignment length:符合比对的比对区域的长度;
mismatches:比对区域的错配数;
gap openings:比对区域的gap数目;
q.start:比对区域在查询序列(query id)上的起始位点;
q.end:比对区域在查询序列(query id)上的终止位点;
s.start:比对区域在参考序列(refer id)上的起始位点;
s.end:比对区域在参考序列(refer id)上的终止位点;
e-value:比对结果的期望值,将比对序列随机打乱重新组合,和数据库进行比对,如果功能越保守,则该值越低;
bit score:比对结果的bit score值;
需要注意的是-db不能只写到存放nt库的目录这一级,后面需要加上库的类型,这里使用的nt库,所以加上nt。输出结果文件类型选择的-outfmt 7 qseqid sseqid evalue pident ppos length mismatch gapopen qstart qend sstart send bitscore staxid sscinames stitle ,这种格式的文件中包含更多的比对及物种信息。
NT数据库官网会定期更新,不同版本的数据库鉴定结果可能会存在差异。NT比对的输出结果只是作为参考,考虑到近源物种等情况,样本是否污染,数据是否可以用,可以与目标物种参考基因组比对后确认,重测序数据推荐使用bwa软件进行比对。
参考文章:
[1] #
[2] #
天津:18710280840/022-24986099
北京:400 1869 509
邮箱:marketing@kangpusen.com
地址:北京市昌平区中关村生命科学园生命园路4号院4号楼7层

图文来源:北京康普森农业科技有限公司










 下载app
下载app