问题分诊室
智因助力复杂的家族性遗传性出血性毛细血管扩张症的基因诊断
发布时间: 2022-06-08
HHT是一类以鼻出血、皮肤和粘膜毛细血管扩张,以及脏器动静脉畸形(AVM)为特征的常染色体显性遗传病。HHT具有遗传异质性,多数为ENG或ALK1(即ACVRL1)基因缺陷导致的HHT1和2型,生长/分化因子2(GDF2)基因杂合性突变导致的HHT被称为第5型(HHT5),以往报导较少,因此对其突变谱以及临床表型的异质性还知之甚少。
HHT发病具有显著的年龄特征,例如儿童期患者毛细血管扩张的表现往往缺如,或仅有轻微鼻衄,因此临床对疑似患儿或HHT患者家系成员,往往要随访至成年才能获得明确的HHT临床证据。现今,基因诊断技术的应用为早期HHT诊断提供了“金标准”,也为HHT临床表型和诊疗,以及发病机制研究均提供了方法和依据。
近日,北京儿童医院呼吸科刘金荣博士、赵顺英教授等研究者在著名儿科杂志PEDIATRICS(IF: 5.4,中科院1区)发表了一篇病例报道,对智因东方团队的贡献表达了致谢。报道为GDF2基因相关的遗传性出血性毛细血管扩张症(HHT)提供了宝贵的临床研究实例。
患儿为5岁2个月男孩,父母为近亲婚配(图1)。临床初诊发现,该男孩主要表现为非综合征性低氧血症,体格检查有杵状指及手指甲床发绀。动脉血氧饱和度(SaO2)仅为82%-86%。肺功能(脉冲示波系统,IOS),肺部增强CT和非造影超声心动图均未发现异常。肺灌注扫描显示大脑和双侧肾脏具有放射性,表明由于肺动静脉畸形而存在肺外分流。根据以上临床发现,患儿诊断为HHT。通过全外显子组测序和突变位点家系成员验证,进一步诊断为GDF2基因纯合变异c.1060_1062delinsAG(p.Tyr354ArgfsTer15)导致的HHT5。该截断突变分别遗传自父母,然而患者家族并无HHT家族史。有趣的是,基因检测进一步发现,患儿7岁的同胞姐姐同样有该GDF2基因纯合变异,但没有临床症状。
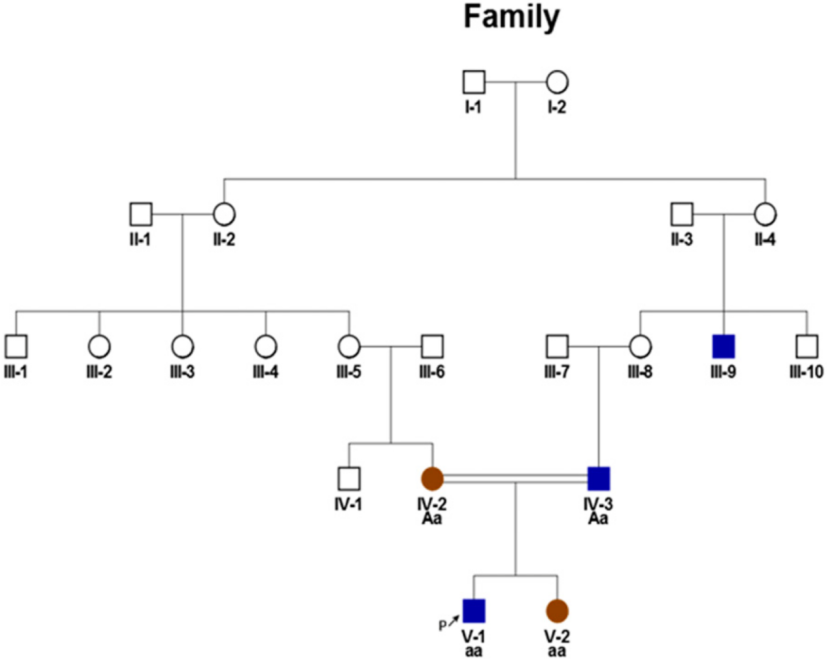 图 1 患者5代家系图
图 1 患者5代家系图
鉴于以上的初步发现,研究者进一步对核心家系成员进行了细致的检测,并仔细探寻了包括先证者在内的五代成员病史。进一步调研发现患者母亲有躯干的一处毛细血管扩张,其姐姐左额部也有同样性状的先天性病损,然而一直被当作痣(图2A);患儿父亲和父亲的舅舅均有幼年时的鼻衄史,后者症状相对较重,持续至成年(图1)。这些发现展示了HHT家族性发病的特征,然而在缺少典型先证者及正确诊断的情况下,家族病史极易被忽略。
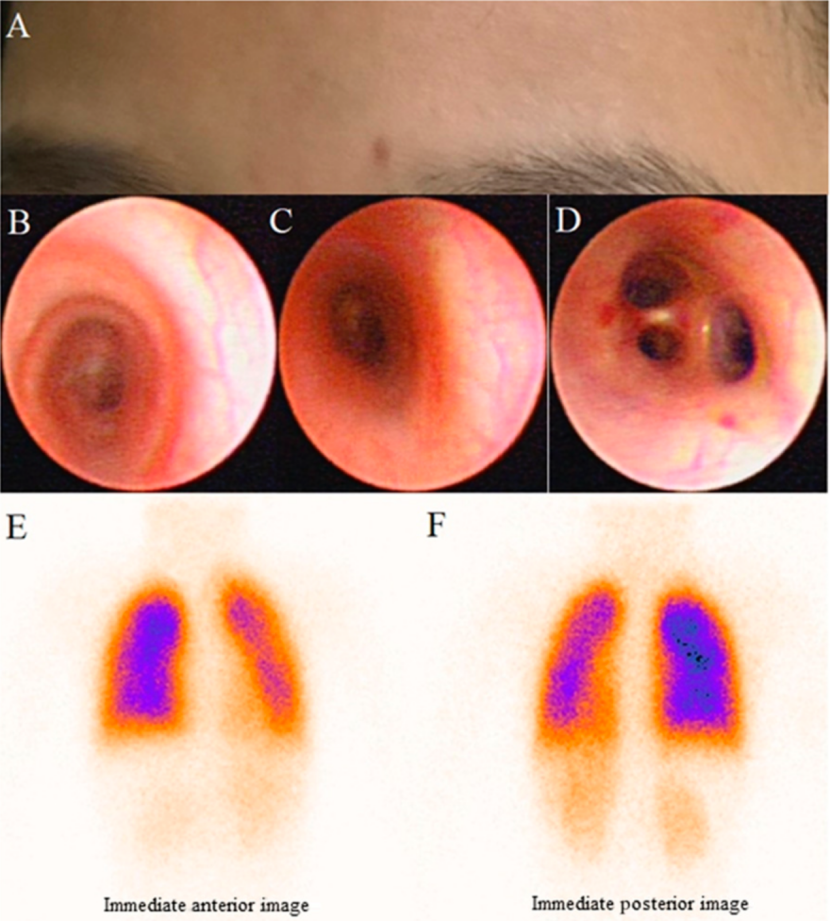 图 2 A. 示患儿姐姐左眉内上部的毛细血管扩张,3 X 2mm。B-D. 支气管镜检查显示小血管轻度增生、支气管壁局部出现粘膜下出血点。E, F,肺通气/灌注扫描示双侧肾脏出现放射性影像,表明存在肺内分流。
图 2 A. 示患儿姐姐左眉内上部的毛细血管扩张,3 X 2mm。B-D. 支气管镜检查显示小血管轻度增生、支气管壁局部出现粘膜下出血点。E, F,肺通气/灌注扫描示双侧肾脏出现放射性影像,表明存在肺内分流。
鉴于患者较重的血氧分压降低,考虑存在肺AVM。通过肺通气/灌注扫描,发现患儿脑及双肾均有显影。由于灌注的放射性分子,例如Tc-99m,大于微血管的直径,因此进入肺动脉的造影分子无法再进入体循环。因此,脑及双肾均有显影表明肺内存在AVM,导致放射性分子溢出到体循环,即存在肺内分流。(图2 E, F)
从该案例的发现推测,GDF2截短突变Tyr354ArgfsTer15可能并未影响GDF2-ALK1的TGF-β信号通路激活功能,然而对ENG-GDF2复合物的形成和功能将造成影响,这也许能够解释这一HHT家系较轻微的HHT表型。
在内皮细胞中,GDF2与I型受体ALK1结合,通过TGF-β信号通路激活下游的SMAD1/SMAD5,从而调控内皮细胞的正常生成。其中GDF2的Ser24和Arg78是所有TGF-β配体的共有保守位点,而Lys64被实验证实是与GDF2-ALK1复合物形成的核心位点,因此在理论上,截短的GDF2蛋白链可能并不影响GDF2介导的TGF-β信号通路激活;同ALK1一样,ENG,一种TGF-β1和TGF-β3的辅助受体,主要与GDF2结合来激活TGF-β信号通路。
GDF2主要通过AA402 - 416蛋白链区域与ENG结合,这提示该区域丢失的截短GDF2将导致ENG-GDF2信号通路的失活,而非GDF2-ALK1介导的TGF-β信号通路。在以往所明确报导的HHT5案例中,GDF2的致病突变主要发生在GDF2-ALK1复合物形成的核心区域,Pro85和Arg68,而一个例外是Arg333位点的错义突变,功能验证表明Arg333突变主要影响GDF2蛋白质的加工和成熟,导致其活性降低。因此,ENG-GDF2功能缺陷能够解释该家系的表型特征,然而有待于进一步的功能验证。
在该病例家系中,作者进行了已知HHT标志物ENG(血清内联素)和VEGF(血管上皮生长因子)的分析。(表1)结果表明VEGF可能与疾病的严重程度不完全相关(缺少HHT特异性),因其在肺炎支原体肺炎患者和其他对照个体中也可能会出现升高,但可用于动态评估治疗反应;ENG在该家系成员中是正常的,这有助于排除ENG缺陷引起的HHT(如上所推测,ENG-GDF2复合物形成/功能障碍,而非ENG本身的缺陷)。对这些建议具有临床参考意义,但需要更多HHT案例的临床研究进一步来证明。
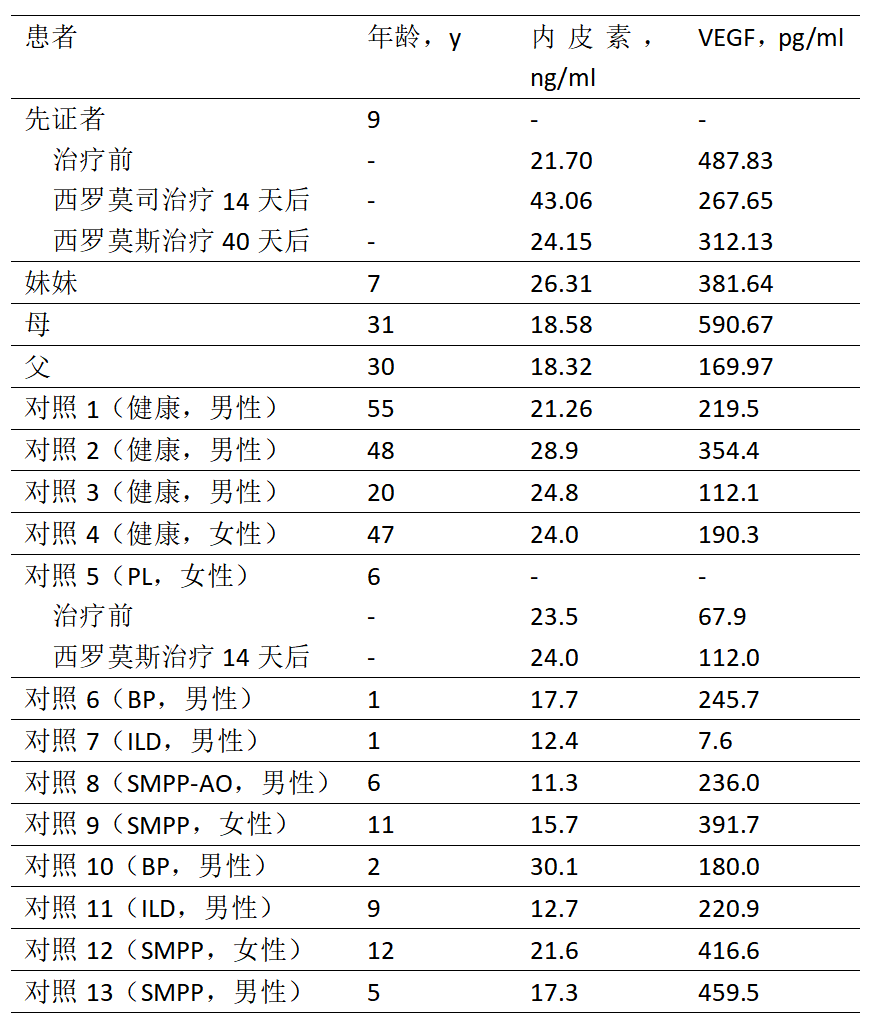 表1 家庭成员血清内皮素和VEGF检测结果。AO:气管闭塞性脉管炎;ILD:间质性肺炎;PL:肺淋巴管瘤;SMPP:重度肺炎支原体肺炎
表1 家庭成员血清内皮素和VEGF检测结果。AO:气管闭塞性脉管炎;ILD:间质性肺炎;PL:肺淋巴管瘤;SMPP:重度肺炎支原体肺炎
该HHT家系成员的临床表型轻微且存在较大差异,即使患儿及其同胞有相同的纯合突变,极大的表型异质性仍使得临床诊断存在诸多疑虑。作者通过细致的家族病史复检和回顾、表型(动脉血低氧分压)的深入探究、基因突变的分子功能分析,以及临床标志物的探索验证,对该HHT家系从表型特征、病理和分子机制,到临床应用进行层层剥茧,展示了一个疑难HHT诊断的优秀范例。
参考文献:Liu J, Yang J, Tang X, et al. Homozygous GDF2-Related Hereditary Hemorrhagic Telangiectasia in a Chinese Family. Pediatrics. 2020;146(2):e20191970. doi:10.1542/peds.2019-1970
文章出自:智因东方微信公众号





 下载app
下载app